

")
Page Contents
Acromegaly Definition
Acromegaly is a syndrome associated with excessive production of growth hormone (GH). In children, the same condition is called gigantism. Acromegaly is an insidious, chronic and debilitating (decreasing strength and vitality) disease associated with tissue overgrowth and increased death chances (mortality).
Causes of Acromegaly
Acromegaly is caused commonly by pituitary adenomas (malignancies of the pituitary gland), usually, a macroadenoma (75%) of cases produce growth hormone. Rarely ectopic tumors may also lead to production of growth hormones or growth hormone releasing hormone (GHRH) and cause this syndrome. Less than 1% are malignant (most are benign). Growth hormone is produced by 20% of pituitary tumors (second most common after hyperprolactinemia 80%).
Growth hormone excessive release occurs most commonly between the 3rd and 5th decades of life (30-50 years) and the following clinical findings are present
Clinical findings in Acromegaly
- Various skeletal and soft tissue changes can be found
- Enlargement of the hands, feet, coarsening of facial features, thickened skin folds.
- Increase in size of worn hats, shoes, gloves and rings
- Increase in size of the tongue
- The nose and mandible (prognathism and separation of the teeth) enlarge, sometimes causing underbite (the lower jaw protrudes more than the upper jaw)
- The voice becomes deeper
- Increase sweating
- Obstructive sleep apnea can also occur (suddenly wake up due to decreased breathing while sleep)
- Internal organs are enlarged (heart, lung, spleen, kidneys and liver)
- Interstitial oedema, osteoarthritis, and entrapment neuropathy (carpal tunnel syndrome) (median nerve is depressed in the snuffbox)
- Menstrual problems are common in women because prolactin is co-secreted side by side by the growth hormone secreting tumour.
- Around 10-20% of patients develop cardiac anomalies such as hypertension, arrhythmias, hypertrophic cardiomyopathy and accelerated atherosclerosis.
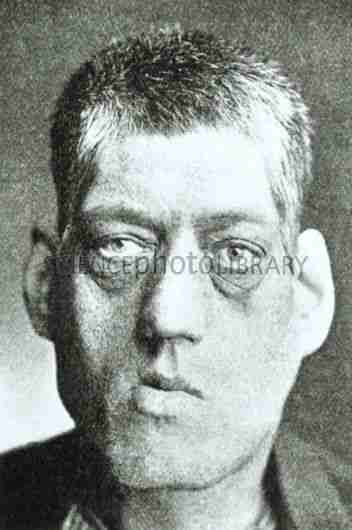
Metabolic changes in Acromegaly
Metabolic changes in acromegaly include impaired glucose tolerance (80%) and diabetes (10-20%). Hypertension is seen in one-third of patients. Headaches and visual field loss may occur also, Articular cartilage in the joints may proliferate and cause problems of joints.
Diagnosis of Acromegaly
Patients with acromegaly have symptoms for an average of 9-10 years before the diagnosis is made because it is a chronic disease. The best initial test is IGF-1 (insulin-like growth factor) level. IGF-1 is produced from the liver along with other hormones due to the action of growth hormone. IGF-1 level if found significantly increased for age-matched normal individuals is a positive screen for acromegaly.
Confirmatory testing involves measurement of GH after 100 g of glucose is given orally as glucose counteracts the GH level (If glucose is high then growth hormone levels should decrease). If GH levels remain elevated (greater than 5nanogram per millilitres) then this suggests acromegaly. Measurement of IGF (somatomedin) correlates with the disease activity.
Radiologic studies such as CT scan and MRI are used to find the location of the tumour Not to find the presence of disease. These tests should be done only after positive findings in the IGF or GH tests. MRI is better than CT scan. MRI will show a tumour in 90% of cases of acromegaly.
Management of Acromegaly
The objective of the management of the disease is to lower the growth hormone levels to normal, stabilize or decrease the tumour size, and get normal pituitary function back. Transsphenoidal (tansverse of the sphenoid bone in the skull) surgery provides a very rapid response. Hypopituitarism can result in 10-20% of cases. Primary treatment in that case is surgery.
Somatostatins (counterparts of somatomedin IGF) are the drugs of choice. Octreotide and lanreotide reduce growth hormone values in about 70% of the patients and also decrease tumour size in 20-50% of the patients as these are somatostatins. Octreotide is the best medical therapy for acromegaly (1st drug of choice). The main side effect of somatostatins is cholestasis (reduced movement of the bile in the gallbladder) which can lead to cholecystits and gall bladder stones.
Dopamine agonists such as cabergoline and bromocriptine may be used if surgery is non responsive as they inhibit the release of anterior pituitary releasing hormones from the hypothallamus.
Pegvisomant is a growth hormone analogue that antagonizes GH by blocking its binding to the GH receptors on the liver and in this way GH is not utilized. Pegvisomant is a second line drug for acromegaly.
Radiotherapy is used only if drug and surgery does not work. This is a slow relieving procces and may result in hypopituitarism in 20% of the patients.
Complications of Acromegaly
Complications of acromegaly may arise from the pressure of the tumour on the surrounding structures as pituitary is present in the sella turcica near the optic chiasm. Also by an invasion of the tumour to the brain or sinuses. Other complications include cardiac failure (due to excessive heart overgrowth and is the most common cause of death in acromegaly patients), diabetes mellitus because growth hormone counter regulates insulin action and insulin is not increased causing hyperglycemia. Cord compression may occur. Visual field defects (most commonly bitemporal hemanopsia) which is due to compression of the optic chiasm and is characterized by loss of looking laterally in both eyes (tunnel vision).
You may also want to look at some other diseases











{kind=link}